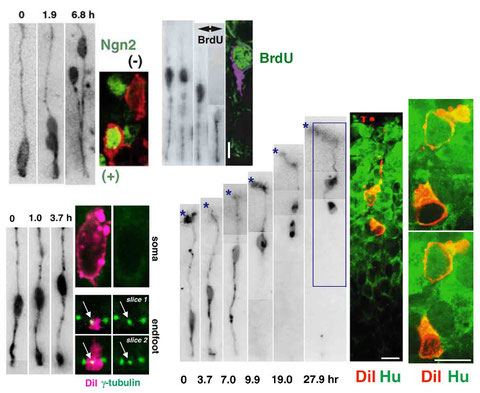







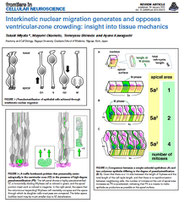



Immunostaining of singly DiI-labeled mouse cortical neural progenitor cells and their daughter cells
(following live observation of these cells in slice culture)
by Takaki Miyata
January 17, 2020
Previously used in:
Miyata, T., Kawaguchi, A., Okano, H., and Ogawa, M. Asymmetric inheritance of radial glial fibers by cortical neurons. Neuron 31, 727-741 (2001)
Miyata, T., Kawaguchi, A., Saito, K., Kawano, M., Muto, T., and Ogawa, M.: Asymmetric production of surface-dividing and non-surface-dividing cortical progenitor cells. Development 131, 3133-3145 (2004)
Miyata, T., and Ogawa, M.: Twisting of neocortical progenitor cells underlies a spring-like mechanism for daughter cell migration. Curr.Biol. 17, 146-151 (2007)
Tamai, H., Shinohara, H., Miyata, T., Saito, K., Nishizawa, Y., Nomura, T. and Osumi, N.: Pax6 transcription factor regulates interkinetic nuclear movement in cortical progenitor cells via centrosomal stabilization. Genes Cells 12, 983-996 (2007)
Miyata, T.: Development of three-dimensional architecture of the neuroepithelium: Role of pseudostratification and cellular 'community'. Dev. Growth Differ. 50, S105-S112 (2008)
Ochiai, W., Nakatani, S., Takahara, T., Kainuma, M., Masaoka, M., Minobe, S., Namihira, M., Nakashima, K., Sakakibara, A., Ogawa, M., Miyata, T.: Periventricular Notch activation and asymmetric Ngn2 and Tbr2 expression in pair-generated neocortical daughter cells. Mol. Cell. Neurosci. 40, 225-233 (2009)
1) Fix DiI-labeled embryonic mouse cerebral walls (coronally sliced, 0.2-0.3 mm thick, cultured in collagen gel) in 4% PFA (in PBS, room temperature, 10 min).
Note: DiI-labeling had been performed before slicing (to the basal/outer surface of unsliced hemispheres freed from meninges).
2) Take each cerebral-wall slice of interest out from the gel and embed it in low melting-temperature agarose (1-2%, in a small box ------ ~1 cm x ~1cm x ~1cm ——— hand-made with aluminum foil), then section (50 micrometer thick) using a vibratome.
Note: Usually several slices were cultured together in a same gel (culture dish). Not to damage each slice during its removal from the gel, I cut the surrounding gel using micro-scissors. To avoid possible denature of antigens to be immunostained, care was taken not to put the slice into too hot agarose (temperature and solidification timing were controlled using ice-containing waters surrounding the aluminum foil box).
3) Collect and transfer vibratome sections (50 micrometer thick, DiI-labeled slice in the center, surrounded by agarose) floated in PBS (no detergent) one by one to 24-well plates (containing PBS) using tweezers. Check them under a fluorescent (inverted-type) microscope as to which vibratome section(s) contained the DiI-labeled cell of interest (a single elongated cell sectioned obliquely can sometimes be separated into two or more sections).
Note 1: During vibratome sectioning, the slice, collagen gel, and agarose can be physically separated, which is troublesome (transfer of slices becomes difficult). For more firmly attaching collagen gel and agarose to the slice part, adding gelatin (1-2%) seemed to be effective. And, too quick vibratome sectioning (horizontal to-and-fro movement and/or forwarding of the blade) may not be a safe method.
Note 2: Frozen section using OCT compound or other related materials removes or diffuses DiI fluorescence, and therefore sporadic (single-cell) DiI labeling cannot use it (even though massive labeling like retrograde labeling of axon bundles would be more resistant).
4) Based on obtaining this 50-micrometer-thick section, one can perform the following immunostaining using a very low but sufficient concentration of Triton X-100 (0.01% in PBS). Primary antibodies were diluted in this buffer (0.01% Triton X-100 in PBS) and treated at 4 centidegree overnight (or longer, if needed/appropriate, up to two overnight).
Note: Using hydrophobic marking ink, prepare a small (5-10 mm diameter) circle on a 35 mm or 60 mm plastic culture dish bottom (like for glass slides before immunostaining of frozen sections) and fill the circle with cold PBS containing 0.005% TritonX-100 (50-100 microliter, this solution will be used throughout the following washing steps). Then, transfer each vibratome section manually to the circle (0.005% TritonX-100 in PBS) under a dissection microscope using a tweezer. Then using 0.2 mL Pipetman under the dissection microscope, the 0.005% TritonX-100 in PBS was removed and the primary antibody solution in 0.01% Triton X-100 in PBS (50-100 microliter) was applied. If you are very unlucky (the cell of interest is exactly ay the center, 25 micrometer from both surfaces, it might be weakly stained or not stained due to insufficient penetration of the primary antibody, otherwise ---- if the cell of interest is within 20 micrometer or less from the surface ---- antibodies will sufficiently reach it). Getting thinner vibratome sections may be difficult: 40 micrometer-thick sectioning seems possible, but much more difficult than at 50 micrometer thick.
5) Next day, remove the primary antibody solution similarly using a 0.2 mL Pipetman under the dissection microscope, and apply and immediately remove the washing solution (0.005% TritonX-100 in PBS) (this “washing” was repeated twice or three times). Then, apply a new solution for secondary antibody(s) (similarly in 0.01% Triton X-100 in PBS). Again overnight (or several hours if we have already experienced that the primary Ab works well) at 4 centidegree (refrigerator).
6) After replacing the 2nd Ab solution with the washing solution in the “well” on culture dish, we transferred (again under the dissection microscope) the immunostaind DiI-labeled slice onto a glass slide (into a drop of the washing solution, 50-100 microliter). After removing the washing solution, apply a semi-viscous-like mounting solution (PermaFluor) and a tiny coverslip (5~7 mm x 5~7 mm, handmade from regular-sized coverslips, not to too flatten or deform the slice due to heaviness of the coverslip) onto it.
Note: Time duration from the applying PermaFluor to immunostaind DiI-labeled section to the confocal microsopic observation was set to become minimal (hopefully within 10 min). It means that mounting was performed one by one. Not simultaneously for multiple different vibratome slices. Slices to be confocal observed were kept most safely in the “well” (in 0.005% TritonX-100 in PBS, on culture dish, in fridge). Polyvinylalcohol or related materials would be bad for DiI again, I guessed, and it seemed to be true.
Please contact Takaki Miyata ([email protected]) if you have questions.